Helminth microbiome study: Helminth Colonization Is Associated with Increased Diversity of the Gut Microbiota
Lee SC, Tang MS, Lim YAL, Choy SH, Kurtz ZD, et al. (2014) Helminth Colonization Is Associated with Increased Diversity of the Gut Microbiota. PLoS Negl Trop Dis 8(5): e2880.
Project Summary
Soil-transmitted helminths colonize more than 1.5 billion people worldwide, yet little is known about how they interact with bacterial communities in the gut microbiota. Differences in the gut microbiota between individuals living in developed and developing countries may be partly due to the presence of helminths, since they predominantly infect individuals from developing countries, such as the indigenous communities in Malaysia examined in this work. The composition and diversity of bacterial communities from the fecal microbiota of 51 people from two villages in Malaysia were compared, of which 36 (70.6%) were infected by helminths. The 16S rRNA V4 region was sequenced at an average of nineteen thousand sequences per samples. Helminth-colonized individuals had greater species richness and number of observed OTUs with enrichment of Paraprevotellaceae, especially with Trichuris infection. A new approach of combining centered log-ratio (clr) transformation for OTU relative abundances with sparse Partial Least Squares Discriminant Analysis (sPLS-DA) was developed, to enable more robust predictions of OTU interrelationships. These results suggest that helminths may have an impact on the diversity, bacterial community structure and function of the gut microbiota.
Resources
The Full manuscript is available at:
Helminth Colonization Is Associated with Increased Diversity of the Gut Microbiota
Soo Ching Lee,Mei San Tang,Yvonne A. L. Lim,Seow Huey Choy,Zachary D. Kurtz,Laura M. Cox,Uma Mahesh Gundra,Ilseung Cho,Richard Bonneau,Martin J. Blaser,Kek Heng Chua,P'ng Loke. May 22, 2014 PLoS NTD DOI: 10.1371/journal.pntd.0002880
Selected Results from the Study
Table 1: Baseline characteristics of the Malaysian indigenous study participants (N = 51).
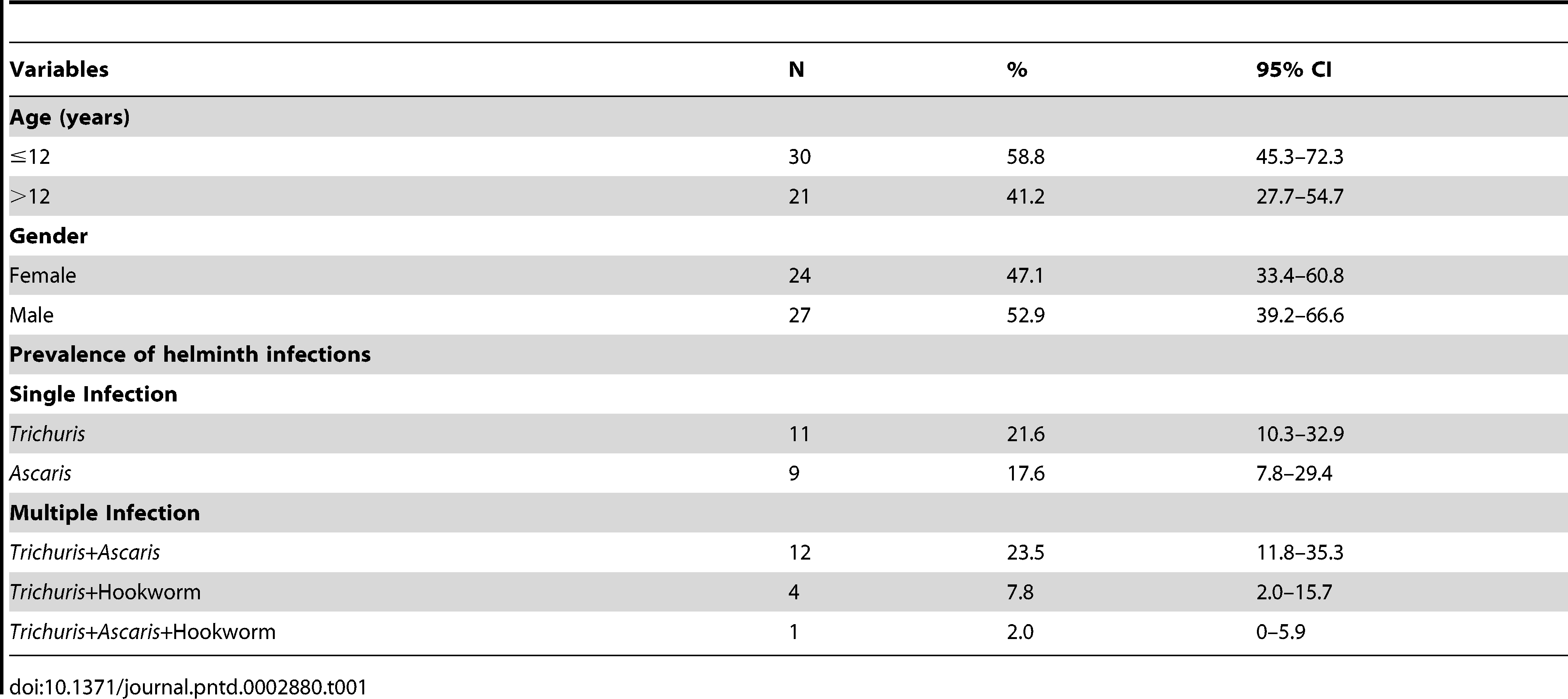
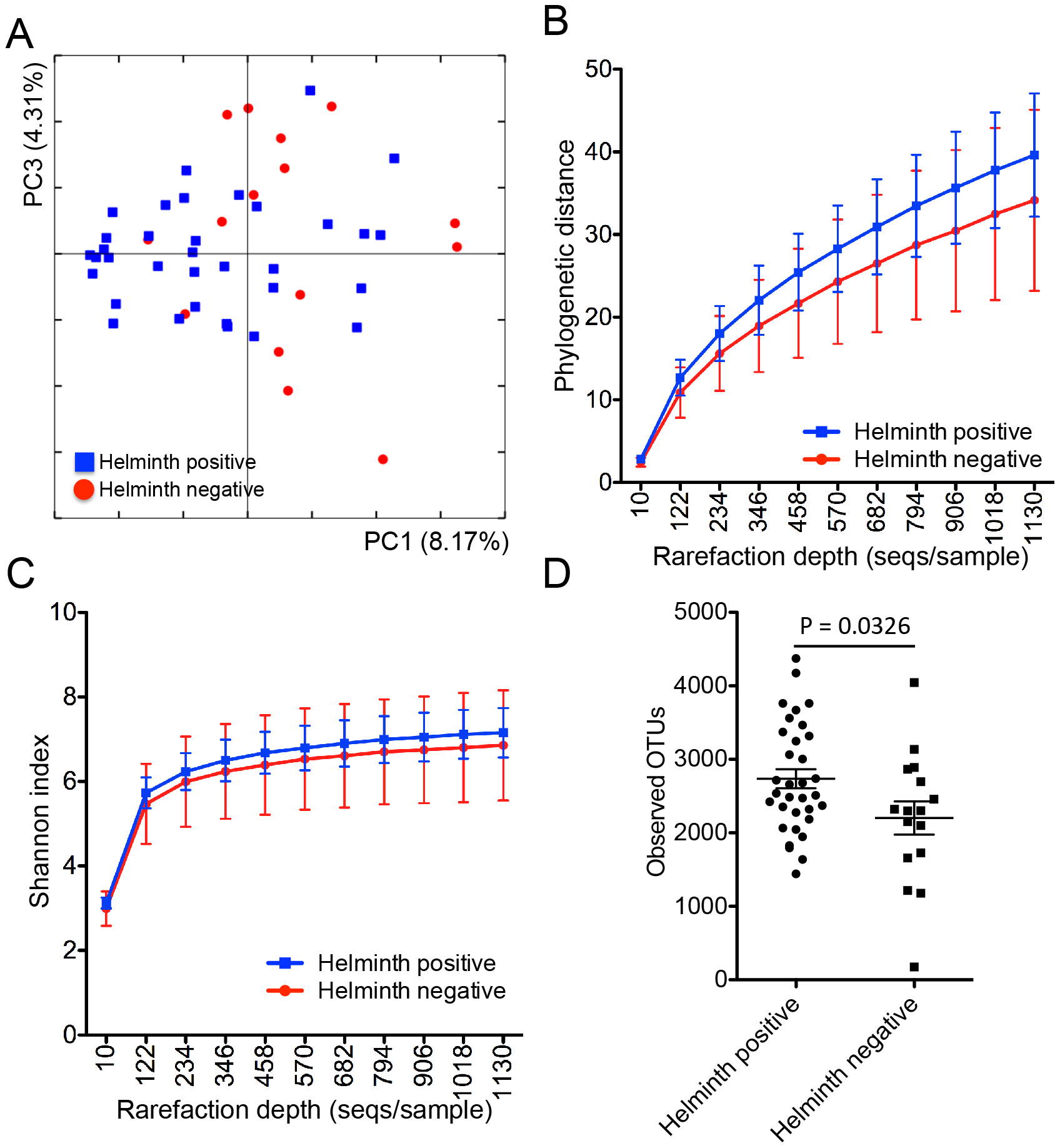
Figure 2. Beta and alpha diversity for the 51 subjects. (A) PCoA of the microbial communities in helminth-positive and helminth-negative samples. Clustering of helminth-positive subjects could be observed, which is statistically significant (p = 0.04). Rarefaction curves calculated for phylogenetic distance (B) and shannon index (C) demonstrating the higher microbial diversity found among helminth positive subjects. (D) total number of observed OTUs in each individual sample was compared between helminth positive and negative groups.
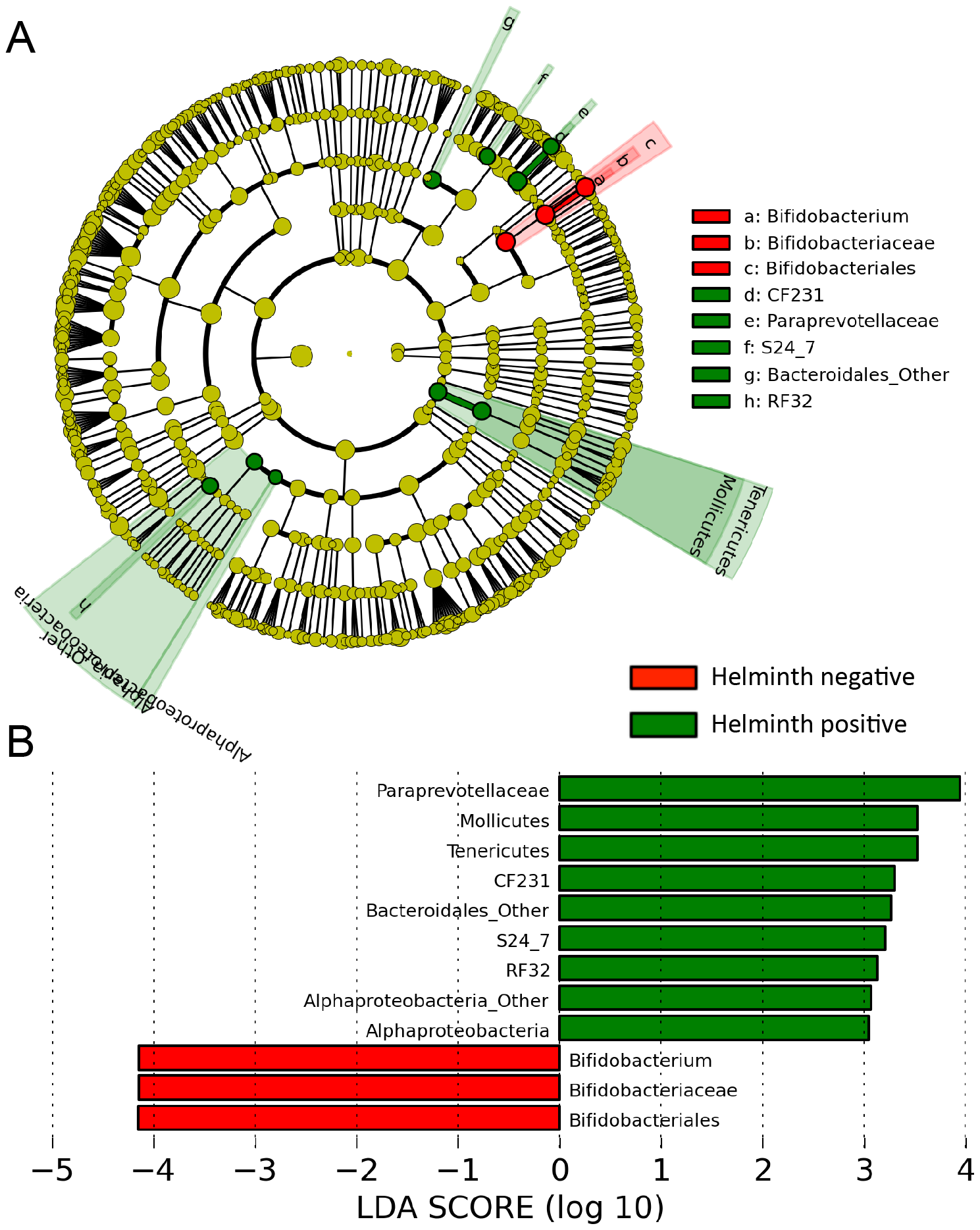
Figure 3. Different abundances of bacterial communities between helminth-positive and negative subjects. With LEfSe for data analysis and visualization key OTUs were identified as differentiating between helminth-positive and helminth-negative fecal samples. (A) Bacterial taxa that were differentially abundant in the gut microbiota profiles of helminth positive and helminth negative subjects visualized using a cladogram generated from LEfSe analysis. (B) With a log LDA score above 3.00, an increased abundance of OTUs was found to be contributed by Paraprevotellaceae, Mollicutes, Bacteroidales and Alphaproteobacteria among helminth-positive subjects, while helminth-negative subjects had increased abundance of Bifidobacterium.
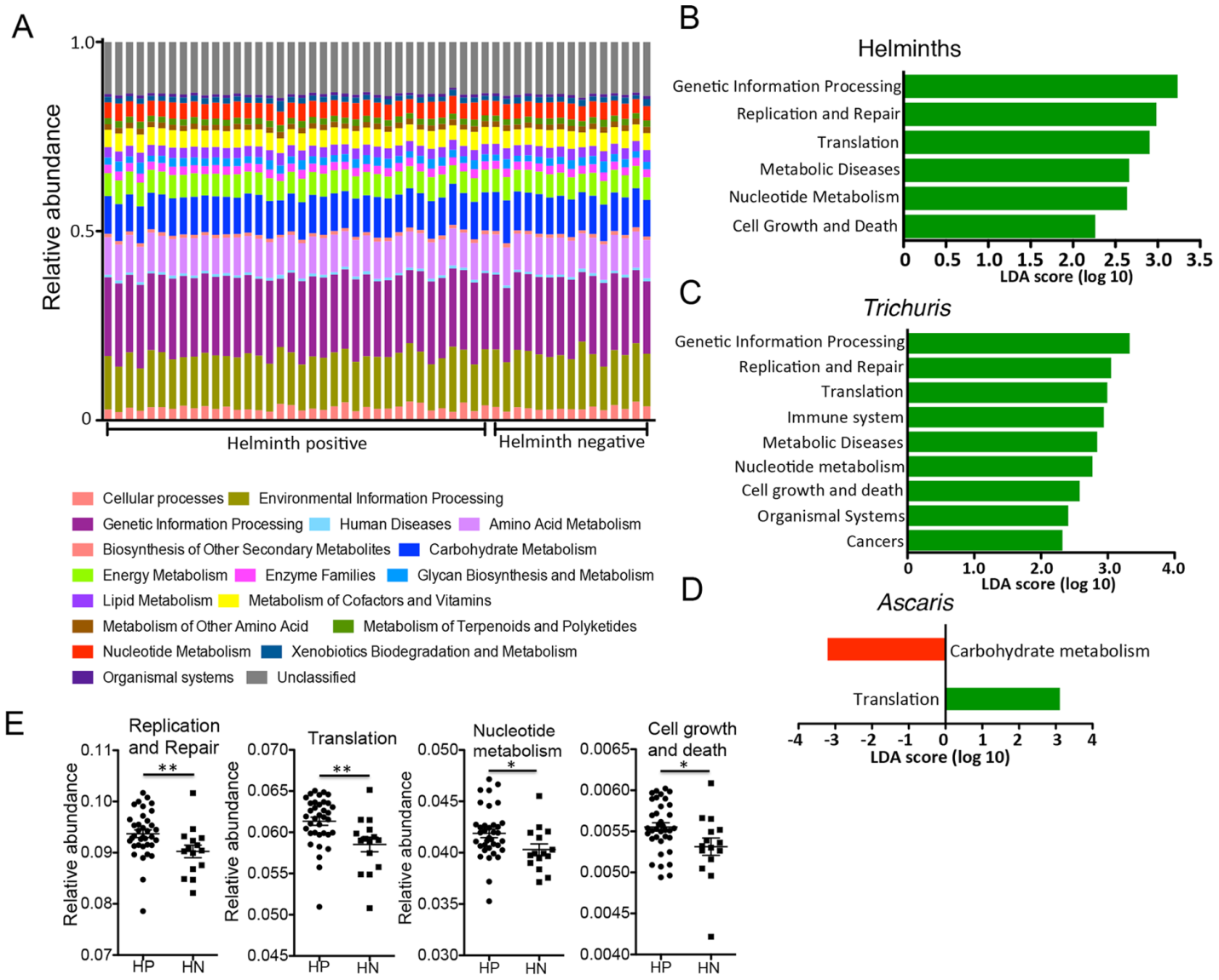
Figure 5. Inferred metagenomic analyses with PICRUSt. (A) Relative abundances of KEGG pathways encoded in the gut microbiota of helminth positive and negative indigenous Malaysians. (B) Supervised comparison using LEfSe identifies differentially abundant KEGG pathways (Log LDA>2.00) in individuals positive for helminths. No functional pathways were differentially abundant in the helminth negative individuals. (C) A similar list of functional pathways was found to be differentially abundant in individuals infected with Trichuris alone. (D) Comparison between individuals who were positive for Ascaris only and individuals who were negative for helminths found translational pathways to be differentially abundant in the former (labeled green) and carbohydrate metabolism pathway to be differentially abundant in the latter group (labeled red). (E) Genes in the pathways of Replication and repair, Translation, Nucleotide metabolism and Cell growth and death are more abundant in helminth positive individuals.

